Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
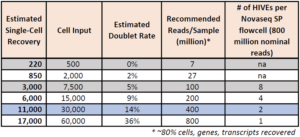
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
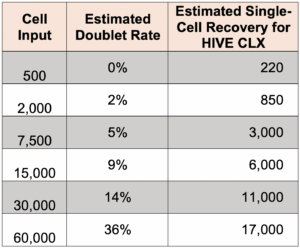
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
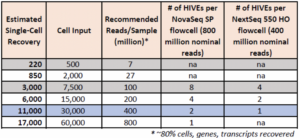
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
Frequently Asked Questions
-
General Information
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
-
Sample Preparation
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
-
Sample Capture
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
-
Storage & Shipping
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
-
HIVE Processing & Library Prep
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
-
QC, Pooling & Sequencing
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
-
Software & Analysis
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.
-
-
What is HIVE scRNAseq?
- HIVE scRNAseq offers a complete solution for single-cell RNA profiling, transforming single-cells to sequencing-ready NGS libraries. The HIVE is a portable, hand-held, single-use device that enables gentle capture, easy storage, and scalable processing for the analysis of single-cell samples.
- A few distinguishing features of the HIVE:
- Integrated sample storage
- Decentralized capture and centralized processing
- Large sample loading volume
- Recovery of fragile cells
- Flexible and scalable workflow
- Strong lysis solution
-
How do I get started with the HIVE?
- We offer three options for getting started: the HIVE Complete Bundle, HIVE Open Source, and HIVE Service
- The HIVE Complete Bundle contains parts and reagents for sample capture, transcriptome recovery, and library preparation. It allows researchers to load their samples and generate sequencing-ready libraries. We also provide our BeeNet analysis software.
- The HIVE Open Source Bundle contains parts and reagents for sample capture and transcriptome recovery. Researchers can use their own library preparation reagents and protocols.
- HIVE Service allows researchers to capture samples and send them to Honeycomb for processing and analysis. This is the easiest way to start for researchers with a low level of NGS experience!
-
Do you provide service?
Yes, we offer HIVE Service as an option for people who do not want to process their own HIVEs. You load the samples, and we do the processing, sequencing, and data analysis. Ideal for new users, pilots, & difficult samples! For more information, click here.
-
What materials are available for training?
- We recommend all first-time users practice with the provided demo parts kit before they begin with their actual samples.
- Users new to working with cells can use the Cell Surrogates to practice cell counting and loading into the HIVE Collector.
- Please contact support@honeycomb.bio to set up a training session.
-
What are the improvements for HIVE CLX versus HIVE v1?
Improvements from HIVE v1:
x4 more cells recovered
Improved workflow and usability
20% more genes & transcripts (from improved chemistry) -
Where can I download the protocols?
Please visit our Support page at honeycomb.bio to download written and video protocols.
-
Are there HIVE-specific requirements for my sample prep?
We recommend whatever sample prep conditions are best suited for your sample type of interest. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA.
-
What do I do if there are clumps in my single-cell suspension?
- If you are concerned about clumps, you can filter your sample through a 70um cell strainer before loading into the HIVE.
- The wash step after cell-loading will help to remove large clumps and debris that do not fit into the 60um wells of the HIVE.
-
How low can my cell viability be?
We recommend aiming for a cell viability >90%. The better the sample quality, the better the data. Samples of 70% viability still generate metrics within a 15% variation of the product specifications.
-
Do you have a recommended protocol for removing red blood cells?
For red blood cell depletion from blood, we recommend using the EasySep™ RBC Depletion Reagent with the EasySep™ Magnet. We recommend processing 0.5 mL of blood at a time and using the magnet 2-3 times to get the best results. After using the EasySep system, we recommend washing the cells twice with cell media + protein to remove platelets and serum protein.
-
What is the shelf-life of my HIVE Collectors?
The HIVE is currently verified for a shelf-life of up to 12-months after receipt when stored at -20C.
-
Are there alternative methods for thawing the HIVE Collectors?
Yes, thawing at 4C overnight is similar to thawing at room temperature for 30 minutes.
-
How long can my HIVE Collector be at room temperature prior to cell-loading?
1 hour, move to 4C if longer than 1 hour.
-
How many cells can be loaded per HIVE?
- For HIVE v1 scRNAseq:
- We recommend loading 15,000 cells per HIVE
- The loading range is 500-30,000 cells per HIVE
- For HIVE CLX scRNAseq:
- We recommend loading 30,000 cells per HIVE
- The loading range is 500-60,000 cells per HIVE
- When counting, please load based on the number of live cells in the sample.
HIVE CLX Cell Loading
- For HIVE v1 scRNAseq:
-
What is the sample loading volume?
- The standard loading method is 1mL of sample (make up the rest of the volume with loading buffer if 1 mL of sample is unavailable).
- The capacity of the HIVE Collector is 4mL.
- If volume is over the 4mL capacity, load sequentially using the spin protocol by removing the supernatant after each spin.
-
What sample types can I use?
HIVE scRNAseq is feasible for eukaryotic cells containing mRNA transcripts with 3’ poly-A tails. Cells should be smaller than 55 µm to fit into the well. We have tested a variety of mammalian cell types, including human filtered blood, PBMCs, mouse splenocytes, and more.
-
Can I use an automated cell counter instead of the disposable hemocytometer?
Yes, but we recommend first doing a comparison with the hemocytometer to ensure that your cell counter is accurate. We recommend the Nexcelom K2 Cellometer.
-
What type of media should I suspend my cells in before loading?
The HIVE is compatible with a broad range of sample buffers. We recommend ~0.1% protein, but up to 10% FBS is okay. We recommend using PBS + 0.1% BSA for cell loading for optimal cell recovery. For HIVE CLX, final cell suspensions must be in media containing at least 1% FBS or 0.1% BSA. An extra wash is recommended for >1% protein.
-
How do you ensure that only one cell is entering each well?
The array is made of picowells that capture cells. Cell capture follows a Poisson distribution. While loading more cells increases single cell recovery, it also increases the rate of doublet formation. The HIVE CLX array features many more picowells than HIVE v1, allowing improved cell loading capacity and single cell resolution.
-
What is the doublet rate?
The following table gives the theoretical multiplate rate (%) for Poisson loading based on the number of cells that were loaded.
-
How long can I store a cell-loaded HIVE at -20C?
HIVE v1 is currently verified for up to 9 months of storage at -20°C, and HIVE CLX is currently verified for up to 3 months. Testing of longer storage periods is ongoing.
-
How should I store my cell-loaded HIVE?
Cell-loaded HIVEs should be frozen. Storing your cell-loaded HIVE at -80C is similar to storage at -20C.
-
Can I go directly from Sample Capture to Processing without storing cell-loaded HIVEs?
Yes, if you are not stopping after Sample Capture, let the cell-loaded HIVEs incubate with the Cell Preservation Solution for 30 minutes before starting the Processing protocol.
-
Why do I have to ship cell-loaded HIVEs with dry-ice?
The Cell Preservation Solution does not fully freeze at -20C. Cell-loaded HIVEs need to be fully frozen (while flat) for shipment.
-
How should I store cell-loaded HIVEs that were shipped to me?
After receiving cell-loaded HIVEs that were shipped on dry ice, we recommend storing them at -80C until processing. For more information, see the receiving instructions in the Sample Loading Protocol.
-
How many samples can I process at once and how long does it take?
Our workflow offers a high degree of flexibility, as each sample is independent. Under normal circumstances, 1 to 24 samples can be worked with in parallel and can be processed and ready for sequencing in 1.5 days.
-
How do I know I have successfully recovered the beads from the HIVE?
After centrifugation, a compact white pellet will be visible at the bottom of the bead collector (more easily seen when holding the spin plate overhead). While removing 300ul, beads will be present in the pipette tip, and the solution will appear cloudy. Afterwards, no pellet will be visible.
-
Can I use a circular 96-well plate magnet instead of a bar magnet?
We recommend a bar magnet for ease of use, but other magnets are acceptable if you’ve successfully used them previously for SPRI clean-up.
-
How do I dispose of my used HIVE parts?
Used HIVE parts can be disposed as non-sharps in standard bio-safety waste.
-
Can the HIVE lysis solution inactivate infectious disease samples?
The lysis solution has been shown to inactivate SARS-CoV-2-infected human whole blood, but we recommend testing your specific samples.
-
What sequencing platform do you recommend?
- HIVE scRNAseq libraries are currently compatible with Illumina sequencing platforms capable of custom dual index paired-end sequencing. We recommend NovaSeq, NextSeq 2000, or NextSeq500/550
- Custom sequencing and index primers are provided with the Library Preparation Reagents
-
How many reads are required for each HIVE?
Required reads can vary depending on number of cells, application, data quality, and sample types.
HIVE CLX
-
Which sequencing kits do you recommend?
- NextSeq 500/550 – 75 High Output cycle kit
- NextSeq 2000- 100 cycle kit
- NovaSeq 6000 – 100 cycle kit
-
What is the recommended sequencing configuration?
- Dual indexed paired-end sequencing with custom sequencing primers
- R1: 25 R2: 50 I1: 8 I2: 8
- The appendix in the Processing User Protocol goes into detail about setting up for sequencing.
-
How can I check my sequencing quality?
- Base quality metrics are used to assess the accuracy of sequencing technology. Illumina provides 2 metrics for assessing base quality: %PF(The percentage of clusters that pass Illumina’s preset filter) and %QC30 (The percentage of bases with a quality score of 30 or higher)
- FastQC can be used to evaluate your sequencing data. FastQC is an open-source quality control tool for high throughput sequence data developed at Babraham Institute. More information and the download can be found at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
How should I demultiplex my samples?
If using the blue CDI Index plate and all indexes used were from the same column of the index plate (for example A1-H1), demultiplex samples based on the index 1 read only, since the index 2 reads will be identical. If the indexes used were from multiple columns or you used the clear UDI Index plate, demultiplex based on the index 1 and index 2 read.
-
When setting up for sequencing, what fragment size should I use?
We recommend using the nominal fragment size of 750 bp for calculating the molar concentration of HIVE libraries when loading Illumina sequencers, even if your fragment analyzer shows a different peak size. Please provide this information to your sequencing facility.
-
What analysis software is provided?
BeeNet™, our software solution specifically designed for HIVE scRNAseq. We also offer Terra users the BeeNetPLUS workflow for end-to-end analysis.
-
What is the difference between BeeNet and BeeNetPLUS?
- BeeNet can be downloaded and used on clusters or cloud via Linux command line. The input file is demultiplexed fastq files and output is a count matrix that can be used for secondary analysis as well as additional QC files.
- BeeNetPLUS is a GUI interface and is hosted on Terra. The input is demultiplexed fastq files and output is an html file that goes through a brief analysis of your data, as well as the standard BeeNet output files.
-
How can I access and use the software?
- Please visit our Support page at honeycomb.bio for instructions on installation and analysis.
- BeeNet can be downloaded and used on clusters or cloud via Linux command line.
- We also host BeeNetPLUS on Terra for users with no command line experience.
-
Can I download example data?
Yes, data is available for download here
-
What is the input file for the BeeNet analysis software?
Demultiplexed Read 1 and Read 2 FASTQ files from Illumina paired-end sequencing.
-
Which reference genome for human do you use?
We have both hg19 and GRhC38 available for use.
-
Can we use our own references?
If there is a reference you are interested in using and it is not available on BeeNet, submit a support request at honeycomb.bio and we can work with you to build a new reference file.
-
Do I need to download references every time?
If you have already downloaded the references and they are still available in the same system, you do not need to download them again.
-
Does the BAM file contain mapping quality information?
Yes it does, however, our BAM files do not contain molecule count information.
-
Do you annotate intronic regions?
Intronic regions are annotated in the BAM file, but they are not included in molecule counts.
-
What types of files does BeeNet generate?
BeeNet produces three types of files: BAM, count matrices, and quality metrics.